
on August 3, 2018
The EMBO Journal published a work by Ilaria Fregno, Elisa Fasana et al. of Maurizio Molinari group, describing a new inducible, receptor-mediated, vesicular transport route between the endoplasmic reticulum (ER), an organelle producing about 40% of the eukaryotic cells proteome, and the lysosomes, the degradative compartments in our cells. This novel intracellular route has been named ER-to-lysosome-associated degradation (ERLAD) pathway and is activated to clear proteasome-resistant aggregated proteins, whose accumulation would be toxic for cells and tissues.
An increasing number of rare, inherited human diseases is caused by protein misfolding. Alpha1-antitrypsin deficiency (ATD) is an example thereof. A point mutation in the alpha1-antitrypsin gene results in production of an aggregation-prone protein in liver cells. In most patients, the mutated protein is efficiently destroyed by ill-defined lysosomal pathways. However, in about 10% of the patients, clearance of aggregated alpha1-antitrypsin is defective. The aggregates are retained and eventually kill the liver cells. This is the major inherited cause of pediatric liver disease and transplantation. Characterize in molecular details how healthy cells efficiently degrade alpha1-antitrypsin aggregates may be instrumental to understand what is wrong in the 10% of ATD patients suffering highly debilitating liver disease.
Available literature ascribes degradation of aggregated alpha1-antitrypsin to autophagy, a major degradative pathway operating in our cells. Autophagy can be strongly induced on administration of drugs such as rapamycin. Unexpectedly however, rapamycin as well as other activators of autophagy are useless, or even deleterious when tested in cellular models of ATD.
This conundrum has now been explained by Molinari’s group in a study that combines the use of confocal laser scanning microscopy, electron microscopy, electron tomography, correlative light electron microscopy, CRISPR/Cas9 genome editing and an innovative imaging technique named HaloTag pulse-chase that allows to fluorescently label and follow over time the intracellular trafficking of newly synthesized proteins-of-interest. Their work showed that degradation of aggregated alpha1-antitrypsin does not require activation of autophagy. Rather, the aggregates are first segregated in ER subdomains, they are then enclosed in transport vesicles that eventually fuse with lysosomal degradative organelles in a complex series of events that has been defined as ER-to-lysosome-associated degradation (ERLAD). The group identified a list of proteins that ensure the efficient clearance of aggregated alpha1-antitrypsin from the ER (i.e., the ER-resident lectin chaperone calnexin, the LC3 receptor FAM134B, the ubiquitin-like protein LC3, various component of the LC3 lipidation machinery, the endolysosomal proteins LAMP1 and RAB7, the ER-resident SNARE protein STX17 and the endolysosomal SNARE protein VAMP8).
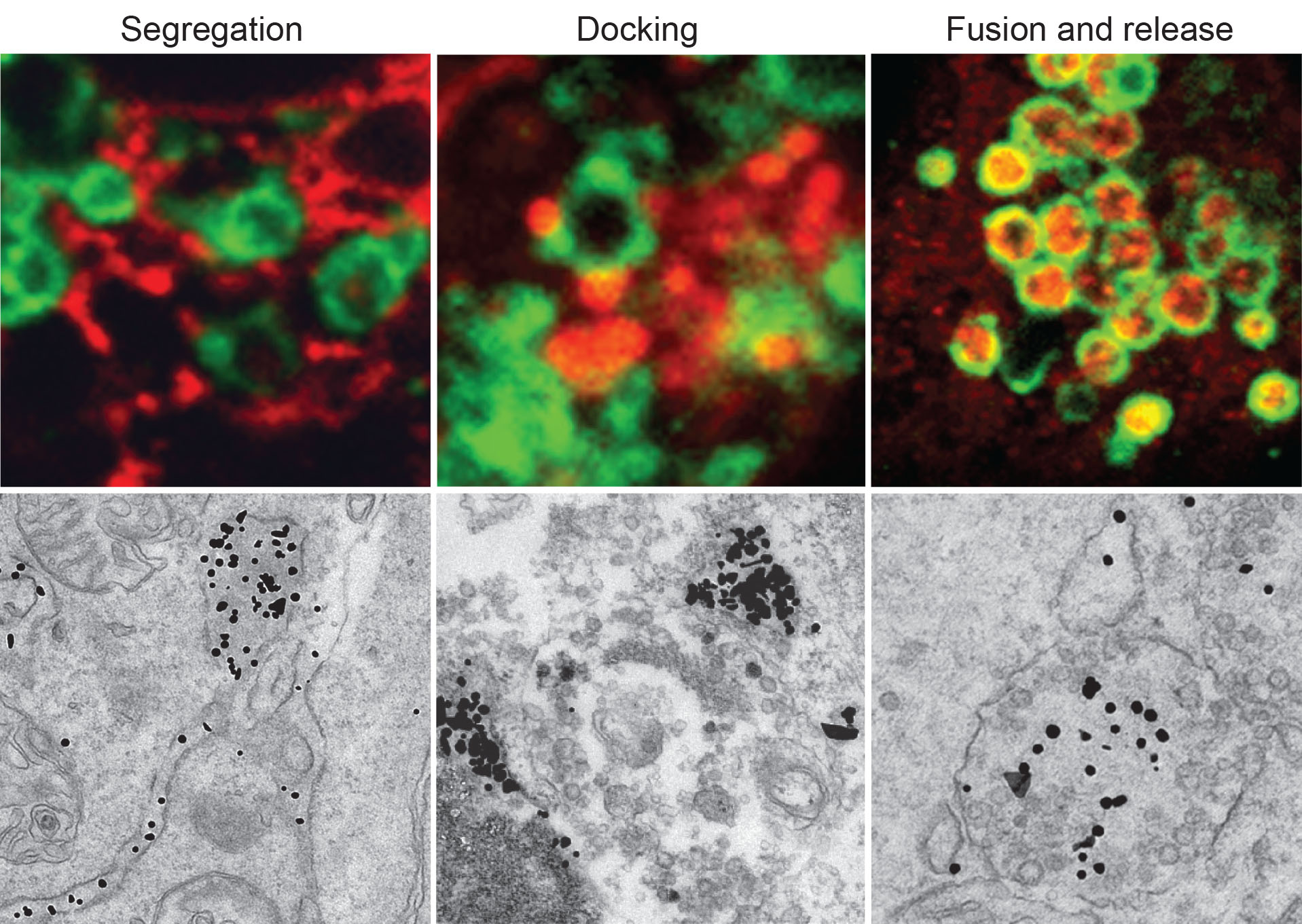 |
| ATZ polymers (red in upper panels and electrondense dots in lower panels) are segregated in ER subdomains before leaving the ER in single-membrane vesicles that dock and fuse with endolysosomes (decorated by Lamp1, Green in upper panels) where ATZ polymers are degraded |
All in all, this work provides new insights in the mechanisms ensuring recognition, delivery and degradation of toxic proteasome-resistant protein aggregates from the ER. Importantly, these results explain the lack of benefits of enhancing autophagy in ATD and, possibly, in other human disorders characterized by formation of protein aggregates in the lumen of the ER. Components of the ERLAD machinery may represent new pharmacological targets for the treatment of gain-of-toxic function diseases causing organ failure.
This work was supported by grants from Alpha-ONE Foundation, Foundation for Research on Neurodegenerative Diseases, the Novartis Foundation, Comel and Gelu Foundations, Swiss National Science Foundation (SNSF).
Article
Fregno I, Fasana E, Bergmann TJ, Raimondi A, Loi M, Soldà T, Galli C, D’Antuono R, Morone D, Danieli A, Paganetti P, van Anken E, Molinari M
doi: 10.15252/embj.201899259